|
TRANSLATE THIS ARTICLE
Integral World: Exploring Theories of Everything
An independent forum for a critical discussion of the integral philosophy of Ken Wilber
  Frank Visser, graduated as a psychologist of culture and religion, founded IntegralWorld in 1997. He worked as production manager for various publishing houses and as service manager for various internet companies and lives in Amsterdam. Books: Ken Wilber: Thought as Passion (SUNY, 2003), and The Corona Conspiracy: Combatting Disinformation about the Coronavirus (Kindle, 2020). Frank Visser, graduated as a psychologist of culture and religion, founded IntegralWorld in 1997. He worked as production manager for various publishing houses and as service manager for various internet companies and lives in Amsterdam. Books: Ken Wilber: Thought as Passion (SUNY, 2003), and The Corona Conspiracy: Combatting Disinformation about the Coronavirus (Kindle, 2020). TABLE OF CONTENTS | REVIEWS
The Subtle Science of Whole Genome SequencingThe Corona Conspiracy, Part 6Frank VisserGenomics has transformed the biological sciences... The genomes of simple bacteria and viruses can be sequenced in a matter of hours in a device that fits in the palm of your hand. The information is being used in a way unimaginable a few years ago. We have seen in the past few episodes 1-5 of this Corona Conspiracy series a lot of disinformation regarding the new coronavirus. Various alternative health celebrities deny either the harmfulness of the new coronavirus—or even its very existence. DISSIDENT VIEWS ON CORONA/COVID-19Various questionable medical arguments have been put forward by these alternative medics/healers/advisors:
Other arguments, of a more technical nature, frequently brought forward are:
And finally it is argued there is a larger, ominous background behind all this:
So there are medical, technical and cultural dimensions to this worldwide Corona pandemic and the various dissident-scientific and/or counter-cultural or conspiracy-related responses it has generated. We will focus on the technical objections here, because first and foremost we need to make sure we really have a real virus in our midst. 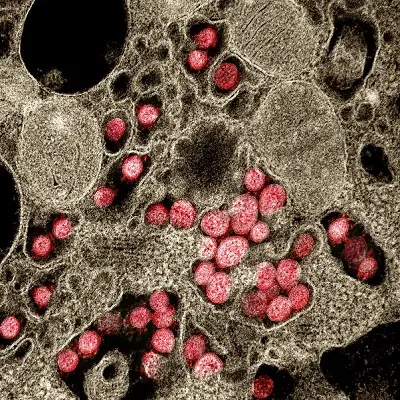
Novel Coronavirus SARS-CoV-2. Transmission electron micrograph of SARS-CoV-2 virus particles, isolated from a patient. (Credit: NIAID)
Do we really have a virus?A good start is one of the first publications about the new virus, published in February 2020 in Osong Public Health Research and Perspectives, a publication of the Korean Centers for Disease Control and Prevention. It states: "This study reports the full genome sequencing of SARS-CoV-2 isolated from putative the 2019 novel coronavirus disease (COVID-19) patients in Korea, by cell culture. The isolated SARS-CoV-2 was named BetaCoV/Korea/KCDC03/2020".[1] This is highly technical material, and I will do my best to understand and report on it correctly, because it touches on two fundamental questions: do we have a new virus, and can we reliably test for it? These are the things the alt-medics such as Stefan Lanka and Andrew Kaufman usually call into question, without providing much details to their readers, to be able to dismiss the contributions of regular virological and genomic science in favor of their own health ideologies. The Korean article contains a few instructive photos and a phylogenetic tree diagram: 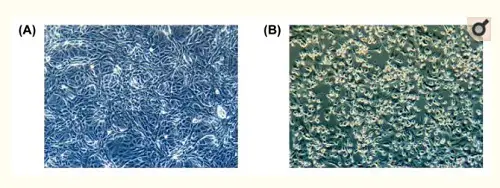
Figure 1: Cytopathic effect of SARS-CoV-2 on Vero cells. (A) Mock inoculated cells (B) SARS-CoV-2 inoculated cells.
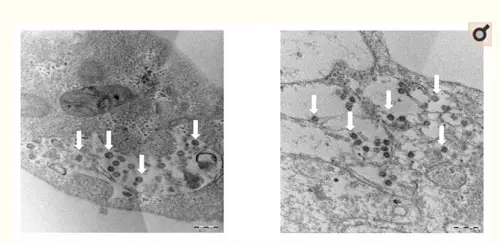
Figure 2: Thin section electron micrographs of Vero cells infected with SARS-CoV-2. White arrows point to aggregates of assembled intracellular virions.

Figure 3: Phylogenetic tree analysis of SARS-CoV-2 based on full genome nucleotide sequences. (A) Gene analysis of SARS-CoV-2 and other coronaviruses, (B) gene analysis of SARS-CoV-2 and BetaCoV/Korea/KCDC03/2020.
The point I would like to make here is: the isolated viral material had an observable pathogenic effect on cells, which the control test did not have. Under a microscope, the viral particles were recognizable as coronaviruses. And the full genome of this particular SARS-CoV-2 viral sample was compared to (1) other coronaviruses (of bats, cows, horses, birds, etc., on the left) and (2) other SARS-CoV-2 genomes (as recently found in China, Singapore, Taiwan, England, the USA, etc., on the right) in the customary phylogenetic tree diagram. And where the photographic material might lead to interpretative difficulties (the major argument of alt-meds), the genomic data are highly specific and meaningful, even painfully precise in their detailedness. Everything under the sun has been genomically sequenced these days: humans, neanderthals, fossil bones, animals, plants, fungi, bacteria, archaea and even viruses. Based on this information the Tree of Life, which currently includes three large domains (Archaea, Bacteria and Eukaryotes), has been fine-tuned to a very high degree. Strange as it may seem, viruses don't really fit in this scheme of all living beings. John Archibald concludes his chapter on "Genomics and the microbial world": Do viruses belong on the tree of life? The jury is still out, but what is inescapable is that viruses are firmly 'plugged in' to the tree of life and capable of shuttling genes from branch to branch. As long as their have been cells on earth, there have been viruses.[2] And perhaps even before there were cells, they infected simple molecular replicators. This may come across as a rather artificial way to establish the existence of viruses. But face it: we know the genomes by now of many visible organisms, from the very large to the very small. That should give us some confidence that the genomes we find for viruses and very primitive archaea, at the very base of the Tree of Life, also refer to real organisms, even if we can't always culture them in the lab, given their extraordinary life styles. Given their small size, smaller than the wavelength of visible light (400-700nm), for a long time analyzing their composition posed a severe challenge to science. Electron microscopy was a huge step forward, but genomics gave us a base-perfect kind of look into their interiors. While their small size might seem a disadvantage, compared to genomes of eukaryotes viruses have less repetitive sequences, which makes their tiny genomes easier to read. Viruses have very compact, straightforward genomes. As a result of this genomic research, it has been found that viruses differ considerably in size. The easiest way to show this is by counting the number of bases (A, C, G or T) they have in their genomes (note: this is not the number of genes, for genes may consist of hundreds of bases):
This image contains 34K information, slightly more than the coronavirus. In sum, we have here quite distinct entities, each with its own characteristic genomes. From genomes we can go to the proteins they code for, and from there to their behavior in the cells they invade. As to the SARS-CoV-2 virus, biologists can read its genome and find areas that specifically code for the spikes or the envelop of the virus. Comparing this genome to other coronaviruses has taught us why this one is particularly harmful. And from that knowledge it becomes understandable that the new coronavirus can cause so many seemingly different types of disease. Do we have reliable tests?This extremely specifc and detailed nature of genomic data also translates into the specificity of virus tests. Instead of just "testing" for "general human genetic material", as the alt-medics usually claim, the tests target specific genetic sequences which are unique to a specific virus only. So let's turn to the second article. Another landmark publication is about how to spot the virus with a so-called Real-Time RT-PCR test, published in January 2020 in Euro Surveillance by a German team. This article claims: "The workflow reliably detects 2019-nCoV, and further discriminates 2019-nCoV from SARS-CoV".[3] What made it possible to act so fast was their long-time experience with SARS-CoV, a similar virus that broke out in 2003, and the international nature of research in the present world. They "only" needed to zoom in on that which makes SARS-CoV-2 unique, a small sequences of bases (for example: GTGARATGGTCATGTGTGGCGG). This missing information was provided by Chinese researchers before the formal release of the full SARS-CoV-2 genome. What we see here is how researchers specifically zoom in on certain small areas of the genome, which contain the genes RdPg (a RNA-dependent RNA polymerase gene), E (envelop protein gene) and N (nucleocapsid protein gene). 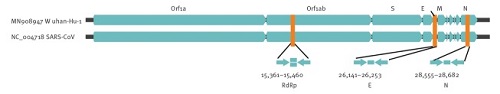
Fig. 4: Relative positions of amplicon targets on the SARS coronavirus and the 2019 novel coronavirus genome
And here you can see where the minimal differences between SARS-CoV-2 and various other viruses could be found (dots represent identical bases, only the base changes are specified where they have been found). They had six samples of the new coronavirus (where only dots can be seen: as to these genes they were identical) and compared it to the earlier SARS-CoV virus, a bat virus from China and a more distantly related bat virus from Bulgaria (where you see more base replacements show up). 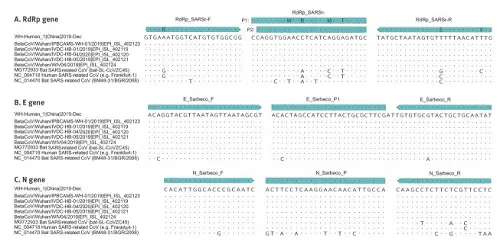
Fig. 5: Partial alignments of oligonucleotide binding regions, SARS-related coronaviruses (n=9)
They applied their test to 22(!) respiratory and other viruses (including MERS-CoV, Influenza, Rhinovirus, Adenovirus and Legionella, to name only a few), and concluded "In total, this testing yielded no false positive outcomes"—meaning, none of these related viruses were mistaken for SARS-CoV-2, which would make the test useless for detecting this virus. The test only reliably detected SARS-CoV-2. Talking about specificity! The authors conclude: "The relative ease with which assays [tests] could be designed for this virus, in contrast to SARS-CoV in 2003, proves the huge collective value of descriptive studies of disease ecology and viral genome diversity." Great insight into how virological science-in-action works is given by "Rapid SARS-CoV-2 whole genome sequencing for informed public health decision making in the Netherlands", which is currently (2020/6/27) only available as preprint on bioRxiv, and is published by a Rotterdam based research group.[4] On February 27th the first COVID-19 case was found in the Netherlands, and two days later(!) the first complete SARS-CoV-2 genome sequences of the first two patients were generated, analyzed and shared. By March 15th, 189(!) full genome sequences were generated and released on GISAID, a worldwide database that contains 55.000 genomic sequences of SARS-CoV-2 submitted from all over the world. This Whole Genome Sequencing (WGS) was accomplished by so-called multiplex PCR for Nanopore sequencing, in which 86 overlapping sequences of 500 base pairs, with 75bp overlap, were used to span the full 30.000bp genome—on a device that can be held in the palm of your hand. This is a stunning scientific and technological accomplishment. The interesting thing is that these relatively cheap and fast devices can directly be put to use in health policy decisions. The tiniest mutations anywhere in the complete viral genome can be detected as it is assembled from patient's samples, and it becomes possible to trace the complex spread of infections across national borders. Assembling the viral genomeAlt-medics such as Kaufman and Lanka often suggest these whole genomes are useless artefacts, because they are based on snippets of RNA that first get multiplied and then arbitrarily "stitched together" with the help of advanced computers to form a digital whole genome sequence, which might as well be related to normal human cell material. This shows a complete lack of understanding of how digital genome assembly works. In the most simple of terms:
A good explanation of how genome assembly works in practice can be found in this video from Bioinformatics DotCa a Canadian Open Source bio-informatics educational institute:
Source: Fundamentals of Genome Assembly, Bioinformatics DotCa.
A more homely example would be: putting multiple copies of a book through a shredder and re-assembling all the pages and sentences in a single, complete book again by comparing the various fragments. Of course, this is a Herculean task no human being would be able to accomplish, but computers can do this ever faster and faster, especially today. With long snippets or "reads" the genome assembly would be easier, but these are very difficult to make. Hence genome sequencing works with relatively small sequences, which can be assembled into a full genome. But the smaller these snippets are, the more challenging the task becomes. Hence the need for high performance computers. 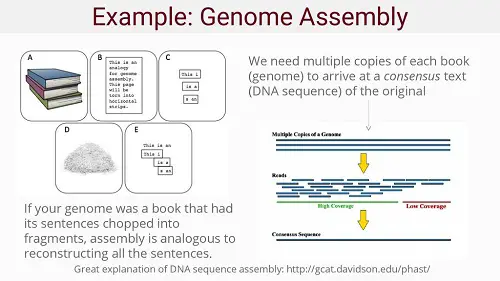
Source: Introduction to Genetics and Genomics (Slide 20).
Here's a final example, of my own making. Say we have several copies of a long sentence, which get cut up in smaller fragments. If the fragments are too small, of the size of individual letters, no sentence assembly will be possible. But if the fragments contain a string of letters, even if the fragments themselves make no sense to us, a computer can assemble the full sentence if it has multiple versions of this fragmented sentence at hand for comparison (in this case there are seven):  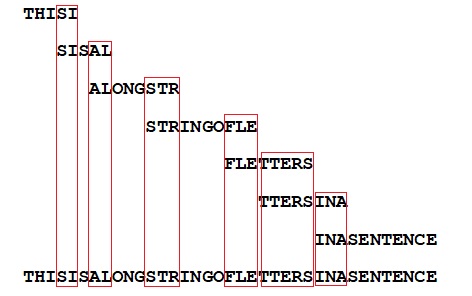
Sentence-assembly based on a set of randomly cut off fragments is feasible if these overlapping fragments have been taken from multiple copies of the sentence.
So we end up here, not with a random string of meaningless fragments "stitched together", but with a meaningful sentence that has a unique structural order. I leave it to you to further explore and read these scientific articles and training videos, to get a feel for the complexity and usefulness of modern genomics. This is how science goes about in knowing viruses to the very base of their genomes.[5] If you want to follow the real-time mutations happening now in the various strands of SARS-CoV-2 that are freely roaming around in the world, do regularly check out the fabulous Nextstrain.org website.[6] Personally I think that dismissing these research data out of hand borders on the insane. Instead of babbling about bubbles in cell photos, Lanka and Kaufman should familiarize themselves with the fields of bioinformatics and genomics. Dismissing these data as nothing more than the observation of exosomes under a microscope (Kaufman), or as the sequencing of mere "dead cell material" caused by the way viruses are cultured during sequencing procedures (Lanka), is without any reasonable scientific ground. You simply don't get this precise and specific knowledge about viruses and their evolutionary relationships based on dead human cell material.[7] Viral biology is an entirely different ball game these days.
Personally I think that dismissing these research data out of hand borders on the insane. Instead of babbling about bubbles in cell photos, Lanka and Kaufman should familiarize themselves with the fields of bioinformatics.
NOTES[1] Jeong-Min Kim et.al., "Identification of Coronavirus Isolated from a Patient in Korea with COVID-19", Osong Public Health Research and Perspectives, 2020 Feb; 11(1): 3-7 [2] John Archibald, Genomics A Very Short Introduction, Oxford University Press, 2018, p. 101. A superb and up-to-date introduction to the wide field of genomics. [3] Victor M Corman et.al., "Detection of 2019 Novel Coronavirus (2019-nCoV) by Real-Time RT-PCR", Euro Surveillance, 2020 Jan 23; 25(3). [4] Oude Munnik, B.B. et.al., "Rapid SARS-CoV-2 whole genome sequencing for informed public health decision making in the Netherlands", bioRxiv, April 25, 2020. Published in: Nature Medicine, 16 July 2020. [5] Ranjit Sah et.al., "Complete Genome Sequence of a 2019 Novel Coronavirus (SARS-CoV-2) Strain Isolated in Nepal", American Science for Microbiology, Microbiology Resource Announcements Mar 2020, 9 (11). [6] Nextstrain, Real-time tracking of pathogen evolution. Nextstrain is an open-source project to harness the scientific and public health potential of pathogen genome data. It provides a continually-updated view of publicly available data alongside powerful analytic and visualization tools for use by the community. Its goal is to aid epidemiological understanding and improve outbreak response. It contains real-time information about the prevalence and spread of SARS-CoV-2, Seasonal Influenza, the West Nile Virus, Mumps, Zika, West-African Ebola, Dengue, Avian Influenza, Measles, Enterovirus and Tuberculosis. Have fun! [7] We will analyse Stefan Lanka's view of viruses and their supposed non-existence in Part 7 of this series.
83 Vaccine Myths from docbastard.net
To all those who claim SARS-CoV-2—or any virus—does not exist: the virosphere consists of 7 realms
11 kingdoms, 22 phyla, 4 subphyla, 49 classes, 93 orders, 12 suborders, 368 families, 213 subfamilies, 3,769 genera, 86 subgenera, 16,215 species. Take that.
https://talk.ictvonline.org/taxonomy/
A summary of early parts of this series has appeared in the Dutch magazine Skepter 33(3), September 2020, as "Viruses don't exist" (covering Parts 1-5). German: Skeptiker (December 2020); English: Skeptic.org.uk (January 2021)
Widget is loading comments...
|
